
RT -PCR即逆转录-聚合酶链反应。原理是:提取组织或细胞中的总RNA,以其中的mRNA作为模板,采用Oligo(dT)或随机引物利用逆转录酶反转录成cDNA。再以cDNA为模板进行PCR扩增,而获得目的基因或检测基因表达。RT-PCR使RNA检测的灵敏性提高了几个数量级,使一些极为微量RNA样品分析成为可能。该技术主要用于:分析基因的转录产物、获取目的基因、合成cDNA探针、构建RNA高效转录系统。
精选百科
本文由作者推荐

RT -PCR
用于检测基因表达水平的反应
中文名
逆转录-聚合酶链反应
外文名
Reverse Transcription-Polymerase Chain Reaction
主要用于1
分析基因转录产物获取目的基因
主要用于2
构建RNA高效转录系统
特点
灵敏而且用途广泛
应用
检测细胞组织中基因表达水平
所属学科
生物学
简介
RT- PCR(Reverse Transcription-Polymerase Chain Reaction)即逆转录PCR,是将RNA的反转录(RT)和cDNA的聚合酶链式扩增(PCR)相结合的技术。首先经反转录酶的作用从RNA合成 cDNA,再以cDNA为模板,扩增合成目的片段。RT-PCR技术灵敏而且用途广泛,可用于检测细胞中基因表达水平,细胞中RNA病毒的含量和直接克隆 特定基因的cDNA序列。作为模板的RNA可以是总RNA、mRNA或体外转录的RNA产物。无论使用何种RNA,关键是确保RNA中无RNA酶和基因组 DNA的污染。
用于反转录的引物可视实验的具体情况选择随机引物、Oligo dT 及基因特异性引物中的一种。对于短的不具有发卡结构的真核细胞mRNA,三种都可。
反转录酶选择
1. Money 鼠白血病病毒(MMLV)反转录酶:有强的聚合酶活性,RNA酶H活性相对较弱。最适作用温度为37℃。
2. 禽成髓细胞瘤病毒(AMV)反转录酶:有强的聚合酶活性和RNA酶H活性。最适作用温度为42℃。
3.Thermus thermophilus、Thermus flavus等嗜热微生物的热稳定性反转录酶:在Mn存在下,允许高温反转录RNA,以消除RNA模板的二级结构。
4.MMLV反转录酶的RNase H突变体:商品名为SuperScript 和SuperScriptⅡ。此种酶较其它酶能多将更大部分的RNA转换成cDNA,这一特性允许从含二级结构的、低温反转录很困难的mRNA模板合成较长cDNA。
合成引物选择
1. 随机六聚体引物:当特定mRNA由于含有使反转录酶终止的序列而难于拷贝其全长序列时,可采用随机六聚体引物这一不特异的引物来拷贝全长mRNA。用此种方法时,体系中所有RNA分子全部充当了cDNA第一链模板,PCR引物在扩增过程中赋予所需要的特异性。通常用此引物合成的cDNA中96%来源于rRNA。
2. Oligo(dT):是一种对mRNA特异的方法。因绝大多数真核细胞mRNA具有3’端Poly(A)尾,此引物与其配对,仅mRNA可被转录。由于Poly(A)RNA仅占总RNA的1-4%,故此种引物合成的cDNA比随机六聚体作为引物和得到的cDNA在数量和复杂性方面均要小。
3. 特异性引物:最特异的引发方法是用含目标RNA的互补序列的寡核苷酸作为引物,若PCR反应用二种特异性引物,第一条链的合成可由与mRNA 3’端最靠近的配对引物起始。用此类引物仅产生所需要的cDNA,导致更为特异的PCR扩增。
试剂准备
1.RNA提取试剂
2.第一链cDNA合成试剂盒
3.dNTPmix:含dATP、dCTP、dGTP、dTTP各2mM
4.Taq DNA聚合酶
影响因素
(一)组织或细胞样本
不是每种组织或细胞都表达所有的基因,即某些基因的mRNA不存在于某些组织或细胞中。因此,确定所选用的材料中是否含有需扩增的目标mRNA模板是实验成败的关键。
(二)引物
合成cDNA第一条链时,引物可用随机6聚核苷酸,或下游引物,或poly(dT),其中以6聚核苷酸的随机引物效果最好。
(三)反转录酶
不同来源的反转录酶均可较好地用于第一链cDNA的合成,其合成受不同RNA模板的影响。提高反转录温度(55℃)、增加1~3倍的酶量可克服RNA二级结构的影响。
(四)cDNA反应产物
合成eDNA第一链所用的RNA模板不影响PCR反应,无需用碱或RNase处理去除。另外,没有必要将cDNA反应产物全部用于PCR,用其1/25~1/10即可。
(五)RNA酶污染
避免RNA酶污染是RT—PCR成功的关键之一。用于RNA操作的各种器皿,包括Eppen—dorf管、Tip头等,都应该用RNA酶抑制剂处理,试剂应该用RNase—free水配制。使用的全部器皿和耐高压的试剂都必须经高压灭菌处理。在操作全过程中,操作者都应该戴一次性手套和口罩,以防污染
操作步骤
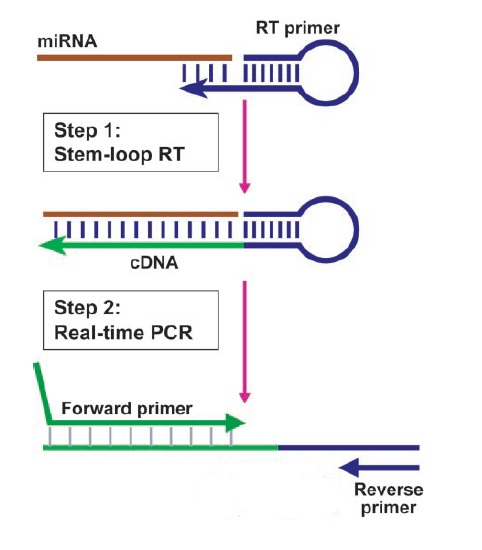
RT-PCR反应原理
1. 总RNA的提取:见相关内容。2. cDNA第一链的合成:目前试剂公司有多种cDNA第一链试剂盒出售,其原理基本相同,但操作步骤不一。现以长沙赢润生物技术有限公司提供的操作手册为例:
(1)在0.5ml微量离心管中,加入总RNA 1-5μg,补充适量的DEPC H2O使总体积达11μl。在管中加10μM Oligo(dT)12-18 1μl,轻轻混匀、离心。
(2)70℃加热10min,立即将微量离心管插入冰浴中至少1min。
然后加入下列试剂的混合物:
10×PCR buffer 2μl
25mM MgCl2 2μl
10mM dNTPmix 1μl
0.1M DTT 2μl
轻轻混匀,离心。42℃孵育2-5min。
(3)加入SuperscriptⅡ1μl ,在42℃水浴中孵育50min。
(4)于70℃加热15min以终止反应。
(5)将管插入冰中,加入RNase H 1μl ,37℃孵育20min,降解残留的RNA。-20℃保存备用。
3.PCR:
(1)取0.5ml PCR管,依次加入下列试剂:
第一链cDNA 2μl
上游引物(10pM) 2μl
下游引物(10pM) 2μl
dNTP(2mM) 4μl
10×PCR buffer 5μl
Taq 酶(2u/μl) 1μl
(2)加入适量的ddH2O,使总体积达50μl。轻轻混匀,离心。
(3)设定PCR程序。在适当的温度参数下扩增28-32个循环。为了保证实验结果的可靠与准确,可在PCR扩增目的基因时,加入一对内参(如G3PD)的特异性引物,同时扩增内参DNA,作为对照。
(4)电泳鉴定:行琼脂糖凝胶电泳,紫外灯下观察结果。
(5)密度扫描、结果分析:采用凝胶图像分析系统,对电泳条带进行密度扫描。
注意事项
1. 在实验过程中要防止RNA的降解,保持RNA的完整性。在总RNA的提取过程中,注意避免mRNA的断裂。
2. 为了防止非特异性扩增,必须设阴性对照。
3. 内参的设定:主要为了用于靶RNA的定量。常用的内参有G3PD(甘油醛-3-磷酸脱氢酶)、β-Actin(β-肌动蛋白)等。其目的在于避免RNA定量误差、加样误差以及各PCR反应体系中扩增效率不均一各孔间的温度差等所造成的误差。
4. PCR不能进入平台期,出现平台效应与所扩增的目的基因的长度、序列、二级结构以及目标DNA起始的数量有关。故对于每一个目标序列出现平台效应的循环数,均应通过单独实验来确定。
5. 防止DNA的污染:
(1)采用DNA酶处理RNA样品。
(2)在可能的情况下,将PCR引物置于基因的不同外显子,以消除基因和mRNA的共线性。
三点注意事项合成cDNA的引物,避免RNA酶的污染,不同来源的逆转录酶。
RT -PCR相关的文章

菊目,拉丁学名Asterales,为显花植物的一目,仅菊科 一科,是显花植物 最大的科。分部广布全世界,热带较少,草本,半灌木或灌木,稀乔木,有乳汁管和树脂道,最显著特点是头状花序,多为一至多年生草本,生长于温带和亚热带的向阳处,分布范围从北极到南极,从海岸到树木线以上的高山。
巴士拉(البصرة,Basra),为伊拉克巴士拉省省会,位于底格里斯河和幼发拉底河交汇的夏台·阿拉伯河西岸,南距波斯湾55公里,是伊拉克第一大港及第二大城。建于635年,曾被战火摧毁,891年被重建。2003年时,全省人口估计约2,600,000人,而巴士拉城则有约1,880,000人。
棕榈 zōng lǘ(学名:Trachycarpus fortunei),又称棕树、唐棕、铁扇棕、鬣葵等,是棕榈属的一种常绿乔木。棕榈广泛栽培于中国、日本、印度、缅甸、美国、欧洲南部等暖温带地区;在中国主要分布在秦岭以南,除西藏外的各省(自治区、直辖市)如广东、云南、上海等。多生长在海拔2000米以
吉尔吉特-巴尔蒂斯坦(乌尔都语:شمالی علاقے,藏语:གིལྒིཏ་བལྟིསྟན),过去称北部地区(شمالی علاقہ جات),当地民族主义分子称为بلاوارستان,位于巴基斯坦控制的克什米尔的北部,是巴基斯坦最北的地区。面积72,496平方公里,2006年人口1,126,
昆仑山脉(英语:Kunlun Mountains)是位于亚洲中部、中国西部的山脉,在地理位置上处于青藏高原北缘,塔里木盆地西南缘、南缘,以及柴达木盆地南缘,西起帕米尔高原东部,东至青海湖西南部的鄂拉山断层;在行政区域上横跨新疆、西藏边界,东延进入青海省内,经纬度在东经74°~101°,北纬32°~3

尚可名片
这家伙太懒了,什么都没写!
作者
